Digital Posters
Spectroscopy: Analysis/Quantification
ISMRM & SMRT Annual Meeting • 15-20 May 2021

Digital Posters
Spectroscopy: Analysis/Quantification
| Concurrent 2 | 15:00 - 16:00 |
2003.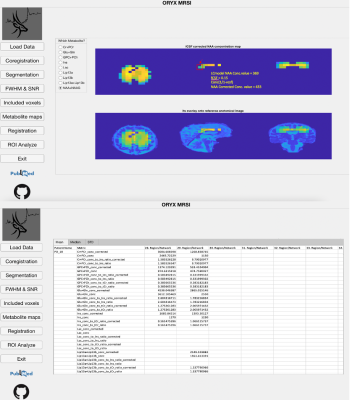 |
ORYX-MRSI: A data analysis software for multi-slice 1H-MRSI
Sevim Cengiz1, Muhammed Yildirim1, Abdullah Bas1, and Esin Ozturk-Isik1
1Biomedical Engineering Institution, Bogazici University, Istanbul, Turkey
Oryx-MRSI is a fully automated software for a comprehensive analysis of multi-slice proton magnetic resonance spectroscopic imaging (1H-MRSI) data. Oryx-MRSI functionality includes chemical shift correction, segmentation, registration, cerebrospinal fluid (CSF) fraction corrected metabolite map generation, registration of metabolite maps onto MNI152 brain atlas, and ROI analysis for metabolite concentration and ratio estimation at brain parcellations defined by resting state fMRI.
|
|||
 |
2004.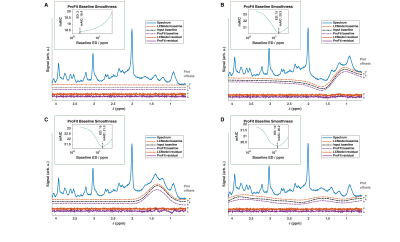 |
ProFit-v3: accuracy and precision evaluation of a new spectral fitting software
Tamas Borbath1,2, Saipavitra Murali-Manohar1,2, Johanna Dorst1,3, Andrew Martin Wright1,3, and Anke Henning1,4
1High-field Magnetic Resonance, Max Planck Institute for biological Cybernetics, Tübingen, Germany, 2Faculty of Science, University of Tübingen, Tübingen, Germany, 3IMPRS for Cognitive & Systems Neuroscience, Tübingen, Germany, 4Advanced Imaging Research Center, UT Southwestern Medical Center, Dallas, TX, United States
In this work, we present the newly developed MRS fitting software ProFit-v3, including adaptive baseline stiffness control and a newly proposed cost function calculation. ProFit-v3 was evaluated for accuracy and precision using both simulated and in vivo spectra, and the results were compared against LCModel. The adaptive spline baseline model of ProFit-v3 modelled different simulated baseline distortions well. The fitting accuracy measured on simulated data was slightly better for ProFit-v3 than for LCModel. While the fitting precision of ProFit-v3 was comparable to that of LCModel for the simulated data, LCModel proved to be somewhat more precise for in vivo spectra.
|
||
2005.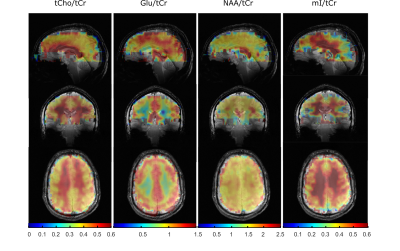 |
Reproducibility and Coverage of Human Whole-brain 1H FID MRSI at 9.4 T after Processing Pipeline Optimization
Theresia Ziegs1,2, Andrew Martin Wright1,2, and Anke Henning1,3
1MRZ, MPI for Biological Cybernetics, Tuebingen, Germany, 2IMPRS for Cognitive and Systems Neuroscience, Tuebingen, Germany, 3Advanced Imaging Research Center, University of Texas Southwestern Medical Center, Dallas, TX, United States
Whole-brain data were acquired using a non-accelerated, non-lipid-suppressed and ultra-short echo time 1H FID-MRSI 2D multi-slice sequence at 9.4 T on human subjects. Data was reconstructed, retrospectively lipid suppressed and fitted including a relaxation corrected macromolecular basis spectrum. Metabolite concentration maps showing expected concentration differences between gray and white matter could be achieved with 89-97% coverage of the whole brain for tCr, tCho, NAA, Glu, and mI. A stack of 32 mm thickness covering the central part of the cerebrum yields anatomically correct maps for Gln, Tau, GABA, NAAG, and GSH. The data acquisition and reconstruction lead to reproducible results.
|
|||
2006.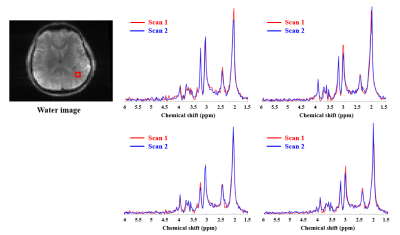 |
Reproducibility of High-Resolution 1H-MRSI at 7T Using SPICE
Pallab K Bhattacharyya1, Rong Guo2, Yudu Li2, Yibo Zhao2, Zhi-Pei Liang2, and Mark J Lowe1
1Cleveland Clinic Foundation, CLEVELAND, OH, United States, 2University of Illinois, Urbana, IL, United States
Subspace-based rapid high resolution MRSI technique SPICE (SPectroscopic Imaging by exploiting spatiospectral CorrElation) was implemented on Siemens Magnetom 7T scanner. Reproducibility of SPICE at 7T with respect to metabolite measures was evaluated for phantom as well as in vivo scans with 3×3×3 mm3 spatial resolution. A phantom and a healthy subject were scanned twice during the same session, with the healthy subject being scanned on two different days. Strong scan-to-scan metabolite concentration correlations were observed in both phantom and in vivo scans. Good reproducibility was also demonstrated from Bland Altman analysis.
|
|||
2007.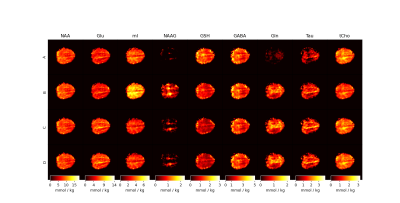 |
Relaxation corrected simulated MM model for improved fitting and quantification of 1H FID MRSI data
Andrew Martin Wright1,2, Saipavitra Murali Manohar1,3, Theresia Ziegs1,2, and Anke Henning1,4
1Max Planck Institute for Biological Cybernetics, Tübingen, Germany, 2IMPRS for Cognitive and Systems Neuroscience, Tübingen, Germany, 3University of Tübingen, Faculty of Science, Tübingen, Germany, 4Advanced Imaging Research Center, UT Southwestern Medical Center, Dallas, TX, United States
Short TE MRS and very short TR (TR < 300) MRSI are popular methods to capture snapshots of the neurochemical profile; however, these popular methods suffer from strong influence from underlaying macromolecular signals. This work shows a simulation method developed at 9.4T and extendable to other field strengths to account for macromolecule signals. The method developed is compared to three more commonly used methods of accounting for macromolecule signals. Results show improved metabolite mapping by use of simulated macromolecule basis vectors.
|
|||
2008.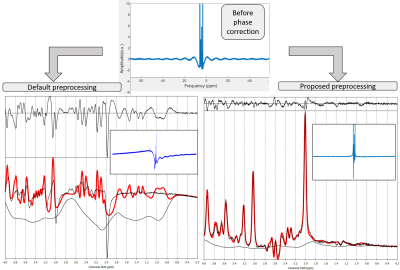 |
Automatic phase order correction in challenging MR spectra
Maria Yanez Lopez1,2
1Centre for the Developing Brain, School of Biomedical Engineering and Imaging Sciences, King's College London, London, United Kingdom, 2Biomedical Engineering Department, School of Biomedical Engineering and Imaging Sciences, King's College London, London, United Kingdom
The aim of this work is to develop an automatic zero and first order phase order correction and apply it to challenging spectra, using an MRS LASER sequence at 3T
|
|||
2009.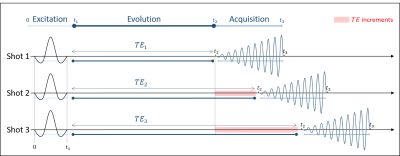 |
Phase Correction in IDEAL-type Rapid Spectroscopic Imaging
Nour EL SABBAGH1, Carine CHASSAIN1, Hélène RATINEY2, Guilhem PAGES1, and Jean-Marie BONNY1
1INRAE, AgroResonance, UR QuaPA, F-63122, Saint-Gènes-Champanelle, France, 2University of Lyon, INSA‐Lyon, Université Claude Bernard Lyon 1, UJM Saint-Etienne, CNRS, Inserm, CREATIS UMR 5220, U1206, F‐69621, Lyon, France
In IDEAL-type sequences, spectral selective pulses are done with an excitation frequency fe dependent on the slice position, and the acquisition is applied with a reception frequency fr. For a proper spectral study, the phase observed during the different evolution times of each shot (TE) has to be dependent only on the CS. A phase coherent fe-fr switch is applied throughout the pulse sequence. However, depending on the frequency commutation position, phase errors are accumulated, disturbing the CS study. This presentation elucidates the impact of the commutation's position and how to correct to resulting phase errors.
|
|||
2010.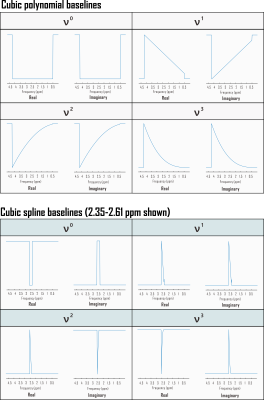 |
Computation of Cramér-Rao Lower Bounds (CRLB) for spectral baseline shapes
Kelley M. Swanberg1, Martin Gajdošík1, Karl Landheer1, and Christoph Juchem1,2
1Biomedical Engineering, Columbia University School of Engineering and Applied Science, New York, NY, United States, 2Radiology, Columbia University Medical Center, New York, NY, United States
Cramér-Rao Lower Bounds (CRLB) are widely applied to characterize the minimum possible variance of metabolite amplitude parameters estimated by linear combination modeling. It has been argued that calculating the CRLB in the absence of baseline terms cannot adequately capture error but that the distribution of spectral baseline modeling parameters themselves cannot be sufficiently represented by this index. In this work we test the practical implications of these principles by treating baselines as linear combinations of polynomials to show that CRLB can under some circumstances offer precision estimates on spectral baseline shapes, notably to the improvement of metabolite CRLB accuracy.
|
|||
2011.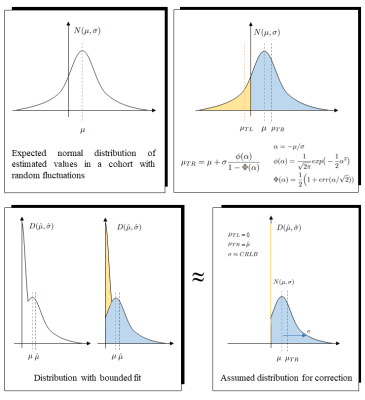 |
Accounting for bias in estimated metabolite concentrations from cohort studies as caused by limiting the fitting parameter space
Rudy Rizzo1 and Roland Kreis1
1Department of Radiology and Biomedical Research, University of Bern, Bern, Switzerland
Potential problems arising from restricting the fitting algorithm in MR spectroscopy to a limited parameter space of physically meaningful values are investigated via Monte-Carlo approach and theoretical considerations. Three parameter-space configurations are compared to evaluate potential bias in the estimated mean cohort concentration for a simulated cohort study typical for conditions for MRS of hippocampus. The bias found for restrictions to positive concentration values can be ameliorated based on an estimate of the distribution width, e.g., based on Cramer-Rao bounds. Loosening parameter space restrictions can also eliminate bias while maintaining the benefit of parameter restrictions for the search algorithm.
|
|||
2012.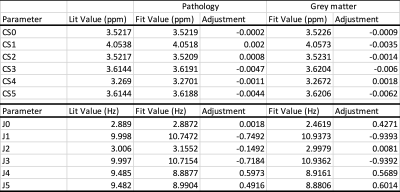 |
Data-driven bases optimization for fitting of in vivo MR spectra
Alexander Saunders1,2 and Stefan Bluml1,2
1Radiology, Children's Hospital Los Angeles/USC, Los Angeles, CA, United States, 2Rudi Schulte Research Institute, Santa Barbara, CA, United States
In vivo MR spectra are commonly analyzed by fitting linear combinations of metabolite basis spectra. In this study we explored whether in vivo spectra themselves can provide information that can be used to further optimize basis spectra. Software was developed that includes the capability to modify basis spectra as part of the fitting procedure. The chemical shifts and J-couplings for myo-inositol were optimized by simultaneous fitting of high-quality spectra.
|
|||
2013.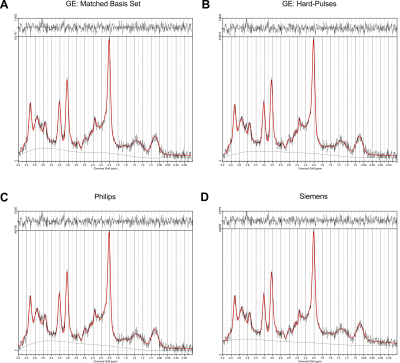 |
The effect of basis sets on the analysis of in vivo brain MRS data obtained with standard PRESS sequences
Martin Gajdošík1, Karl Landheer1, Kelley M. Swanberg1, Lawrence S. Kegeles2,3,4, Dikoma C. Shungu5, Camilo de la Fuente-Sandoval6,7, and Christoph Juchem1,4
1Department of Biomedical Engineering, Columbia University, New York City, NY, United States, 2Department of Psychiatry, Columbia University College of Physicians and Surgeons, New York City, NY, United States, 3New York State Psychiatric Institute, New York City, NY, United States, 4Department of Radiology, Columbia University Medical Center, New York City, NY, United States, 5Weill Cornell Medicine, New York City, NY, United States, 6Laboratory of Experimental Psychiatry, Instituto Nacional de Neurología y Neurocirugía, Mexico City, Mexico, 7Department of Neuropsychiatry, Instituto Nacional de Neurología y Neurocirugía, Mexico City, Mexico
Point resolved spectroscopy sequence (PRESS) is the most commonly used sequence for in vivo magnetic resonance spectroscopy. While implemented by all major vendors, implementation details like timings, durations and shapes of the RF pulses differ among them. Here, we investigate the impact that inappropriate basis information can have on MRS metabolite quantification with linear combination modeling for quantification.
|
|||
2014. |
Bayesian deep learning-based 1H-MRS of the brain: Metabolite quantification with uncertainty estimation using Monte Carlo dropout
HyeongHun Lee1 and Hyeonjin Kim1,2
1Department of Biomedical Sciences, Seoul National University, Seoul, Korea, Republic of, 2Department of Radiology, Seoul National University Hospital, Seoul, Korea, Republic of
Recently, deep learning showed its potential in the quantification of metabolites from 1H-MRS brain spectra. However, previously used standard convolutional neural networks (CNNs) do not provide measurement uncertainty. We investigated the Bayesian CNNs (BCNNs) with Monte Carlo dropout sampling for metabolite quantification with simultaneous uncertainty estimation. The high correlations between the ground truth errors and the BCNN-predicted uncertainty for the majority of the metabolites found in this study may support the potential application of the proposed method in deep learning-based 1H-MRS of the brain for metabolite quantification with simultaneous uncertainty estimation.
|
|||
2015.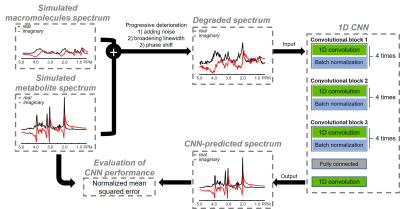 |
Impact of training size on deep learning performance in in vivo 1H MRS
Sungtak Hong1 and Jun Shen1
1National Institute of Mental Health, National Institutes of Health, Bethesda, MD, United States
Deep learning has found an increasing number of applications in MRS. Nevertheless, few studies have addressed the impact of training data size on deep learning performance. In this work, we used density matrix simulation to generate a very large training dataset (70,000 spectra). Then comprehensive comparison was performed to evaluate deep learning performance with different training data sizes.
|
|||
2016.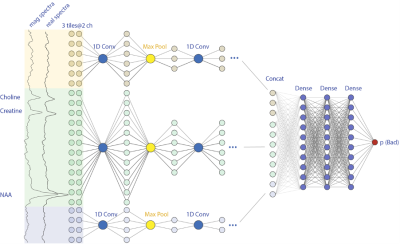 |
Application of Deep Leaning Model for Quality Control of Short-echo 7T MRSI with Various Disease Types
Huawei Liu1, Emily Xie1, Helene Ratiney2, Michael Sdika2, and Yan Li1
1Department of Radiology and Biomedical Imaging, University of California, San Francisco, San Francisco, CA, United States, 2Univ. Lyon, INSA-Lyon, Université Claude Bernard Lyon 1, UJM-Saint Etienne, CNRS, Inserm, Lyon, France
In this project, we adapted the model with various inputs combinations, such as using different tiles, including tissue information and magnitude spectra as additional channels. These variant models were trained and tested in a comprehensive cohort of in-vivo 7T MRSI datasets of various patient types. In our test, an AUC of 0.966 was consistently achieved for the multiple type datasets.
|
|||
2017.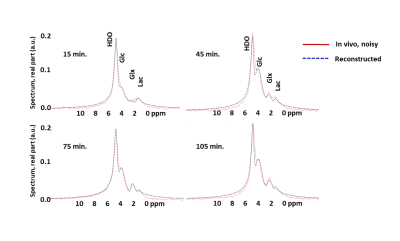 |
Deep Learning Using Synthetic Data for Signal Denoising and Spectral Fitting in Deuterium Metabolic Imaging
Abidemi Adebayo1, Keshav Datta2, Ronald Watkins2, Shie-Chau Liu2, Ralph Hurd2, and Daniel Mark Spielman2
1Mechanical Engineering, Stanford University, Stanford, CA, United States, 2Radiology, Stanford University, Stanford, CA, United States
Deuterium metabolic imaging, a promising tool to probe in vivo glucose metabolism, is severely limited by SNR due to the low gyromagnetic ratio of 2H and the low concentration of metabolites. Recent advances in machine learning techniques to reduce noise is a promising option but obtaining training datasets with good SNR requires prohibitively long scan times. In this work we show that an autoencoder network trained using only synthetic data can reduce noise and provide a good spectral fit for in vivo 3T spectra obtained from human brain after ingestion of deuterated glucose.
|
|||
2018.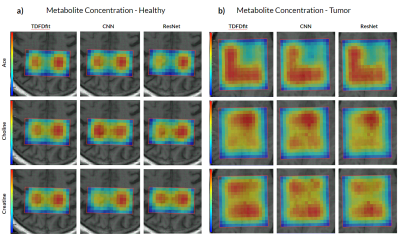 |
Deep Learning Based MRS Metabolite Quantification: CNN and ResNet versus Non Linear Least Square Fitting
Federico Turco1, Irena Zubak2, and Johannes Slotboom1
1Institute of Diagnostic and Interventional Neuroradiology / SCAN, University Hospital Bern and Inselspital, University Bern, Bern, Switzerland, 2Neurosurgery, University Hospital Bern and Inselspital, University Bern, Bern, Switzerland We present and compare two different deep CNN architectures, and VGG-like and an ResNet. We aim at performing metabolite quantification, and compare their performance to a NLLS-fitting algorithm (TDFDfit). We show the performance of the two AI algorithms in a set of 2 in vivo cases as well as in a brain tumor patient. We found that our ResNet outperform the CNN when predicting spatial distribution of metabolite concentration and showed a bigger correlation with the metabolites predicted by NLLS-fitting algorithm. |
|||
2019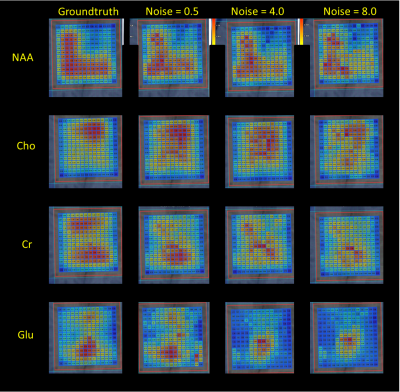 |
Quantification of 2D-MRSI Datasets using Random Forest Regression Comparing to Prior Knowledge Based Spectral Fitting applied to Brain Tumors Video Permission Withheld
Brigitte Schweisthal1, Federico Turco1, Raphael Meier1, Irena Zubak2, and Johannes Slotboom1
1Neuroradiology / Support Center for Advanced Neuroimaging (SCAN), University Hospital and Inselspital, University Bern, Bern, Switzerland, 2Neurosurgery, University Hospital and Inselspital, University Bern, Bern, Switzerland
Robust spectral quantification is essential in clinical 1H-MRSI. A machine-learning technique to quantify 2D-MRSI data aiming to mimic prior-knowledge fitting of the data of glioma patients at 3T. A Random-Forest Regression method was applied on MRSI-data aiming at obtaining improved starting values for the NLLS-algorithm. Enhanced starting values can bring significant developments in the spectral fit quality in clinical 1H-MRS. Different noise levels were compared in order to verify and improve the fitting. Results indicate that this novel approach could increase fitting precision and eliminate possible errors caused by the using uniform starting values and improve method for MRSI-data quantification.
|
|||
2020.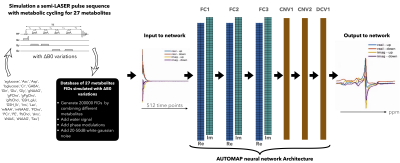 |
In vivo Cerebellum MRSI reconstruction by domain-transform manifold learning
Neha Koonjoo1,2,3, Adam Berrington4, Bo Zhu2,3,5, Uzay E Emir6,7, and Matthew S Rosen2,3,5
1Department of Radiology, A.A Martinos Center for Biomedical Imaging / MGH, Charlestown, MA, United States, 2Harvard Medical School, Boston, MA, United States, 3Department of Physics, Harvard University, Cambridge, MA, United States, 4Sir Peter Mansfield Imaging Centre, School of Physics and Astronomy, University of Nottingham, Nottingham, United Kingdom, 5Radiology, A.A Martinos Center for Biomedical Imaging / MGH, Charlestown, MA, United States, 6School of Health Sciences, Purdue University, West Lafayette, IN, United States, 7Weldon School of Biomedical Engineering, Purdue University, West Lafayette, IN, United States
The recent advances of machine learning in MRSI have mainly been focused on predicting metabolite concentrations and denoising the metabolite-only spectra. Here, we present a deep neural network based on the AUTOMAP formalism to reconstruct metabolic cycle FIDs into the spectral domain. A density matrix formalism was used to generate up/down fields of 1H FIDs of 27 metabolites. B0 inhomogeneity was also included in the simulations. Non water-suppressed up/down field FIDs were fed to the trained network and the proposed reconstruction strategy was validated on simulated FIDs at different noise levels and on an in vivo cerebellum dataset at 3T.
|
|||
2021.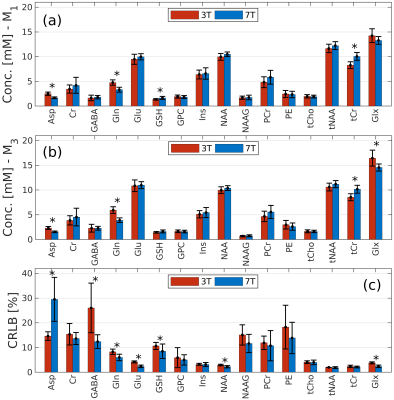 |
On the quantification of the striatum neurochemical profile using STEAM MRS: a comparison of 3T versus 7T in a cohort of elderly subjects
Ana Gogishvili1,2, Christopher E. J. Doppler3,4, Ezequiel Farrher1, Aline Seger3,4, Michael Sommerauer3,4, Ketevan Kotetishvili2, and N. Jon Shah1,5,6,7
1Institute of Neuroscience and Medicine 4, Medical Imaging Physics, Forschungszentrum Jülich, Jülich, Germany, 2Engineering Physics Department, Georgian Technical University, Tbilisi, Georgia, 3Institute of Neuroscience and Medicine 3, Forschungszentrum Jülich, Jülich, Germany, 4University of Cologne, Faculty of Medicine and University Hospital Cologne, Department of Neurology, Cologne, Germany, 5Institute of Neuroscience and Medicine 11, Forschungszentrum Jülich, Jülich, Germany, 6JARA BRAIN Translational Medicine, RWTH Aachen University, Aachen, Germany, 7Department of Neurology, Faculty of Medicine, RWTH Aachen University, Aachen, Germany The primary aim of this study is to quantify the neurochemical profile of the human striatum in vivo in healthy subjects, using data acquired with a single voxel STEAM MRS sequence at 3T and 7T in a case-control study on Parkinson’s disease. Given that the voxel sizes at both fields were different, we intend to ascertain the conditions under which the neurochemical profiles at 3T and 7T are comparable. To this end, we assessed different quantification methods and strategies, including correction for transverse and longitudinal relaxation and for differences in grey and white matter metabolite concentrations. |
|||
2022.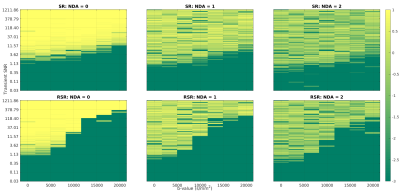 |
The influence of spectral registration on diffusion-weighted magnetic resonance spectroscopy ADC estimates.
Christopher W Jenkins1
1CUBRIC, Cardiff University, Cardiff, United Kingdom
Spectral registration is a powerful frequency and phase offset correction method. Diffusion-weighted MRS (dMRS) provides a particularly stern challenge for drift correction due to the lower SNR, varying lineshape, and increased gradient-induced frequency drift. Here, simulated data are used to examine spectral registration and its new robust iteration in the context of dMRS. The accuracy of these methods is examined across a broad range of SNR, and the effect they have on ADC estimates, investigated.
|
The International Society for Magnetic Resonance in Medicine is accredited by the Accreditation Council for Continuing Medical Education to provide continuing medical education for physicians.