Andrew Martin Wright1,2, Saipavitra Murali Manohar1,3, Theresia Ziegs1,2, and Anke Henning1,4
1Max Planck Institute for Biological Cybernetics, Tübingen, Germany, 2IMPRS for Cognitive and Systems Neuroscience, Tübingen, Germany, 3University of Tübingen, Faculty of Science, Tübingen, Germany, 4Advanced Imaging Research Center, UT Southwestern Medical Center, Dallas, TX, United States
1Max Planck Institute for Biological Cybernetics, Tübingen, Germany, 2IMPRS for Cognitive and Systems Neuroscience, Tübingen, Germany, 3University of Tübingen, Faculty of Science, Tübingen, Germany, 4Advanced Imaging Research Center, UT Southwestern Medical Center, Dallas, TX, United States
A novel method to simulate macromolecule signals to improve MRSI metabolite mapping and quantification with very short TR 1H-FID MRSI. The method
developed is compared more commonly used methods of accounting for macromolecule
signals. Results show improved metabolite mapping.
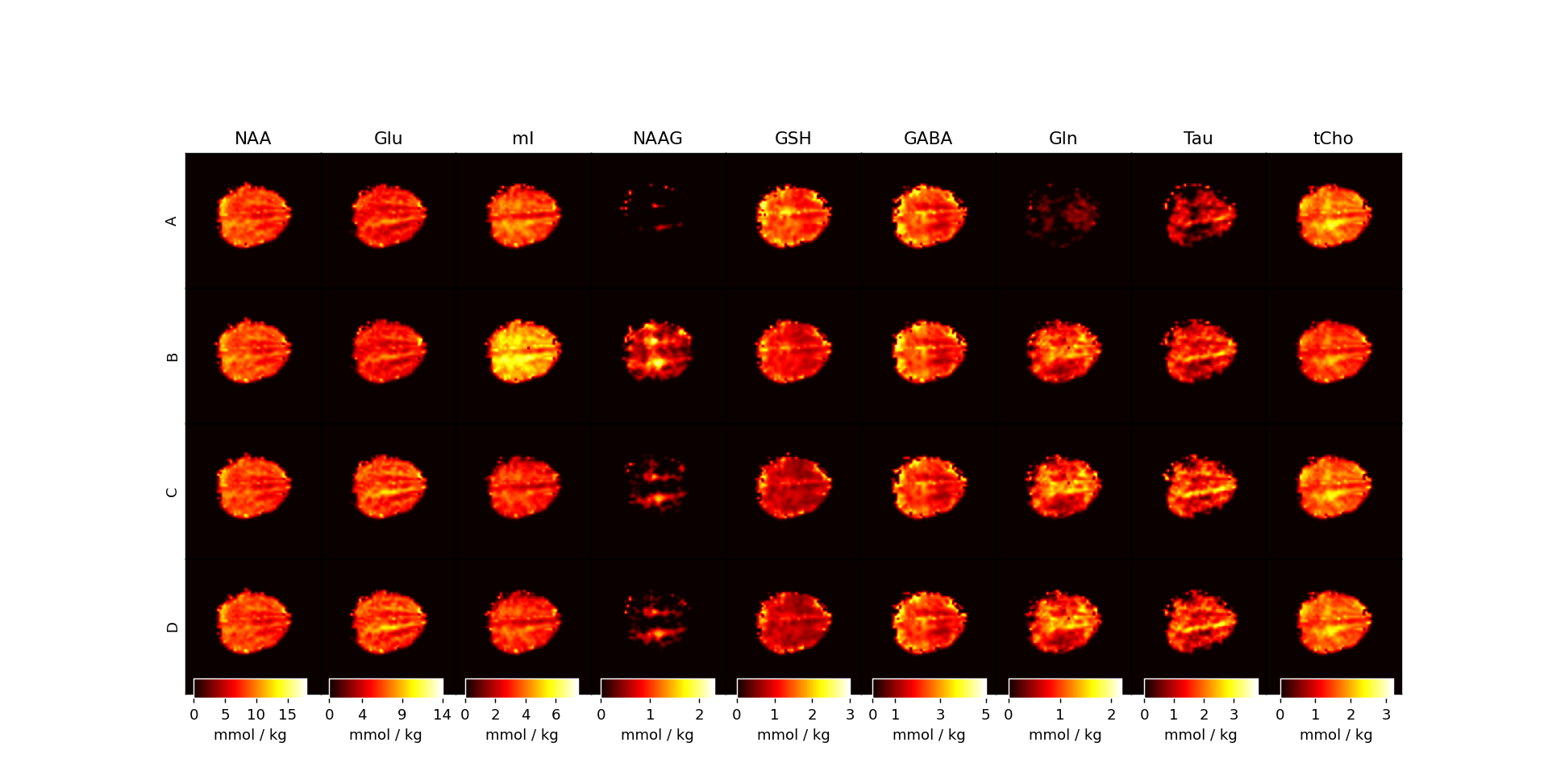
Figure 4: Metabolite maps for the four approaches used for fitting MRSI data. Maps are reported with T1-relaxation corrections and in units of mmol / kg. It is apparent that using simulated MM basis vectors performs best generally when considering the poor fits of NAAG, mI, and Gln from Approach A and Approach B.
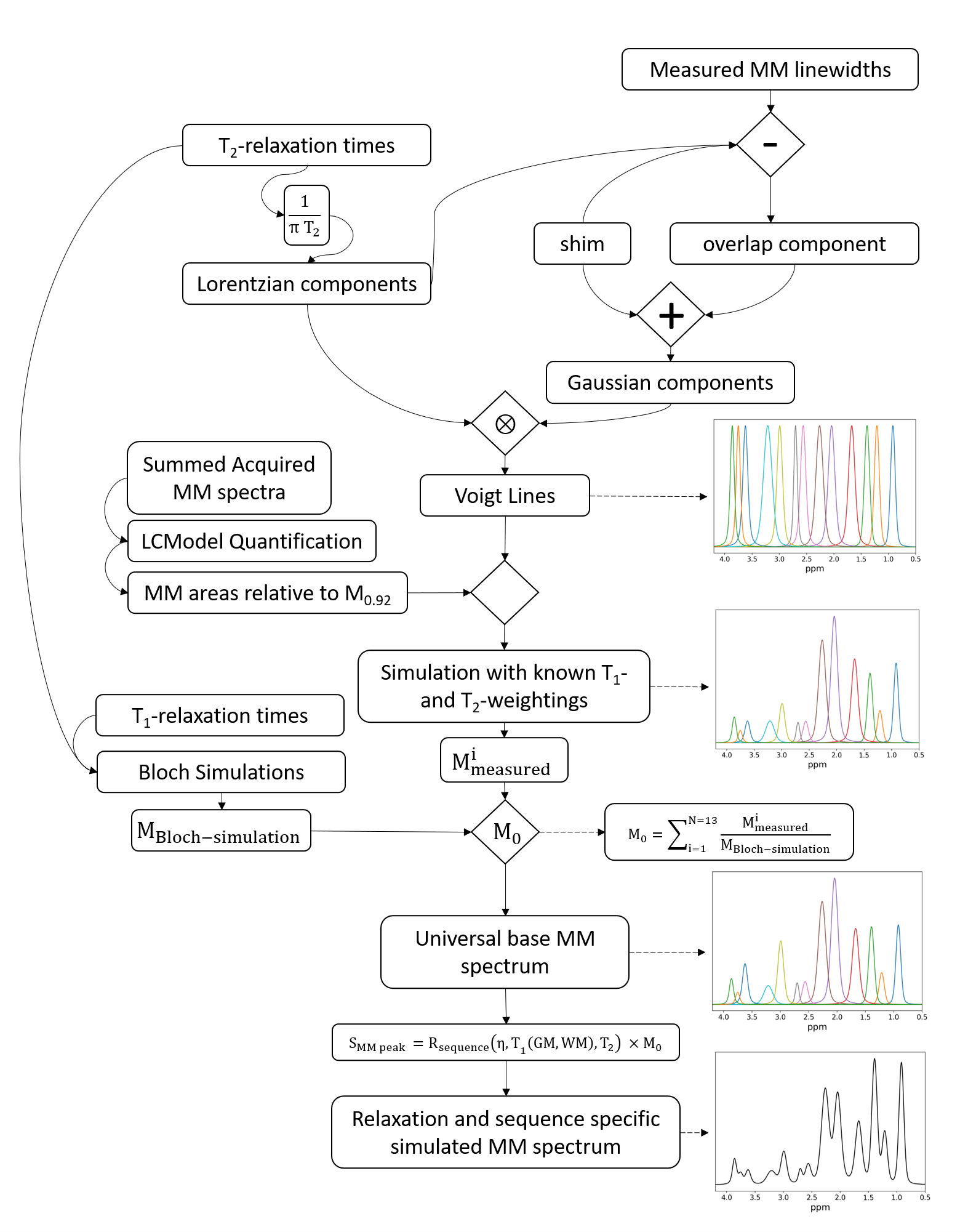
Figure 1: The
relaxation-corrected, sequence-specific MM simulation model algorithm diagram. Voigt lines are simulated using measured Gaussian and Lorentzian lineshapes. Voigt lines are then scaled by measured concentrations of MM from 9.4T and further processed with single-spin Bloch simulations to simulate a universal base MM spectrum which is then attenuated by sequence specific relaxation effects to yield a sequence specific MM basis vector.